Gaussian input file .com or .gjf
To calculate the charge transfer integral between two molecules you will need the minimized atomic orbital coefficients calculated from quantum chemical calculations. If using Gaussian you can tell the program to output this information using the following keywords:
nosymm punch=mo iop(3/33=1)
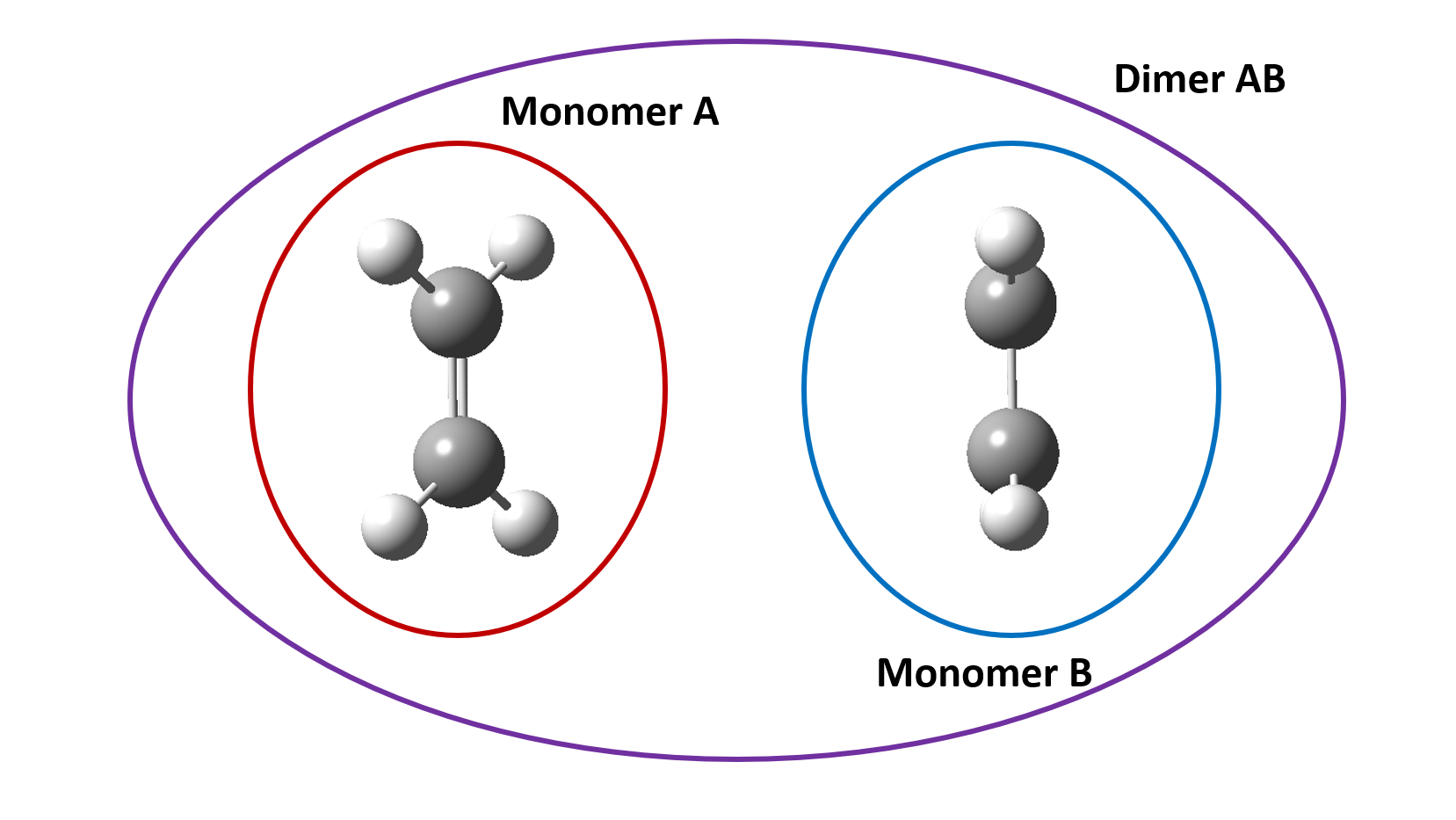
Three separate calculations are needed to calculate the charge transfer integral of a dimer system each of them should contain the keywords shown above.
- Calculation of dimer system consisting of monomer 1 and monomer 2
- Calculation of monomer 1
- Calculation of monomer 2
Both monomer 1 and 2 calculations should remain in the same positions as they appear in the dimer calculation. After the calculation has completed you should have generated the correct output files.
Note
-
In previous versions of
calc_Jit was necessary to ensure the atom labels and positions in the gaussian monomer input files (.gjf) files appeared in the same order as they did in the dimer input files (.gjf). This was to be sure that the basis functions associated with the atoms in the dimer calculations were consistent with the basis functions of the atoms in the monomer calculations. This is no longer necessary if the Gaussian calculations are run with thepop=fullkeyword. The keyword outputs the arrangement of the basis functions in the .log files allowingcalc_Jto automatically line up the appropriate basis functions with the appropriate atoms. A word of warning, if you do not use the keywordpop=full, you must pay special attention to the input coordinates in the .gjf files but also to the coordinates in the .log files. Gaussian will sometimes rearrange the atoms after reading them in to make the computation more efficient, however this will also result in misalignment of the basis functions between dimer and monomer calculations. Fortunately, if you use the keyword IOp(1/22=1) you can force Gaussian to use the coordinates in the order you specified in the .gjf files. -
If you receive an error, it may be the result of differences in the number of AO or MO printed to the fort.7 files. If the number of molecular orbitals is not consistent between the two monomer calculations with the dimer calculation it might be the case that the Gaussian program is simply omitting them to save on the computational cost. However, all orbitals are needed in this calculation. To include them you can add the keyword:
int=nobasistransform
Gaussian output files
Fort.7
The fort.7 file contains the coefficients for each molecular orbital. To run with calc_J, the fort.7 files must be relabeled with a (.pun) extension.
.log
The .log file should contain information related to the overlap matrix which is needed to calculate the transfer integral.